
Biology is being revolutionized by technological advances in DNA sequencing that have made copious amounts of DNA sequence available. To make sense of this deluge of data, biologists will oftentimes “align” the DNA sequences so that they can be readily compared. These comparisons are important for homology detection, recognition of evolutionarily important sites, and a number of other biological endeavors. This process works fairly well for a small number of sequences, but has been unable to maintain its quality with the large number of sequences now available.
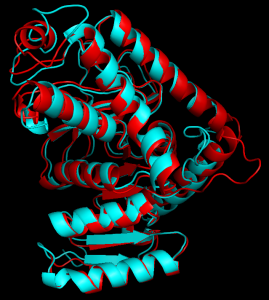
In a recent study, Erik Wright, a graduate student in the Systems Biology theme at WID, investigated potential solutions to the problem of rapidly generating large and accurate biological sequence alignments. He found that scalable alignments can be made by harnessing structural predictions during the alignment process. Although the primary sequence (ACTG…) diverges greatly between organisms, its corresponding 2D and 3D structure is often highly conserved. By aligning the structures and sequences simultaneously, large alignments of diverse sequences can be created that maintain similar accuracy to small alignments. This discovery will enable biologists to better respond to the rapid increase in new DNA sequences.
You can find the full paper, published in BMC Bioinformatics, here.
You must be logged in to post a comment.