Connecting the Dots: a New Method to Understand Cell Type Transitions
Every cell in your body contains the same DNA sequence – the same instruction manual for creating proteins, new cell types, and tissues – and yet you are made up of many different types of cells that perform different functions. Different cells, as it happens, are reading different parts of the instruction manual. Variety in cell types is to a large extent due to epigenetic, rather than genetic, differences – changes that result in different genes being expressed in different cell types.
The lengthy strands of DNA inside the nucleus of a cell are wound around special protein complexes called nucleosomes made up of histone proteins. Histones can be modified by chemical marks, which make it easier or more difficult for other proteins to access and read specific regions of DNA, or genes. Because gene expression depends on histone states, all genes are not expressed at once; genes can be turned on or off.
Rupa Sridharan
Rupa Sridharan, assistant professor of cell and regenerative biology at WID, studies the epigenetics of cell fate. By exploring histone modifications and changes in gene expression across cell types, Sridharan can better understand how cells transition from one type – like a pluripotent stem cell capable of becoming any kind of cell – to a fully differentiated end-type capable of just one function, such as a skin or liver cell. Then, she can reverse the process to create induced pluripotent stem (iPS) cells: cells that were once differentiated, but have been reprogrammed to have their ability to become other cell types restored. Such cells can be used by scientists in regenerative medicine to create new tissues without the need for embryonic stem cells.
Measuring histone modifications to better understand and improve cell reprogramming is not new, but scientists are currently missing a big part of the gene regulatory picture. “People profile histone modifications in different cell types all the time, but the important information that always gets lost is if the cell types are connected,” says Sridharan. For example, two cell types can be derived from the same cell, “but the fact that these cells are connected is information that is lost if we treat each cell type and its corresponding histone modifications as independent of each other.”
The information connecting two related cell types is important because it informs the construction of hierarchies – or cell lineages – and the gene regulatory networks that describe how cell types and their corresponding histone modifications are related. To capture this information, Sridharan partnered with assistant professor of biostatistics and medical informatics Sushmita Roy, a colleague at WID who develops statistical computational methods to identify gene regulatory networks.

Sushmita Roy
“Right now researchers in stem cell biology, like Rupa Sridharan, want to know about cell type specification and how one cell type changes from one state – like a pluripotent state – to another, like a skin cell,” says Roy. “So far people have developed computational methods to look at one cell type independently or pool them together without taking into account how they are related. What we really needed was a way to somehow maintain the individual identity of these cells, but also model the dynamic process of how the histone state changes from one cell type to the other.” Such a method would allow researchers to identify key genes that change their histone state from one cell type to another as the likely drivers of the transition process and to assemble gene regulatory networks that describe cell differentiation.
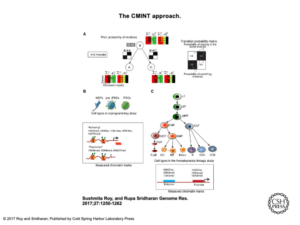
In order to make comparisons across cell types that take individual cell identities and their relationships into account and parse the multitudes of histone modifications and their endless possible combinations, Sridharan and Roy developed an approach called CMINT: Chromatin Module Inference on Trees, where “trees” are hierarchies of related cell types. “Chromatin” refers to the measurements of histone states while “modules” are groups of genomic regions with similar histone states. By searching for regions that change their module membership from one cell type to another, they can begin to identify drivers of cell fate. “We can observe how the dynamics of the chromatin change at the individual gene level,” says Roy.
“Both of us learned something we wouldn’t have without this collaborative project.”
–Rupa Sridharan
Both Sridharan and Roy describe their partnership as a truly interdisciplinary venture encouraged by the Wisconsin Institute for Discovery’s collaborative culture that brought together complementary skills from both sides. “Both of us were engaged in it at an intellectual level, 50/50. We had a lot of discussions, a lot of brainstorming sessions,” says Roy. Sridharan adds, “both of us learned something we wouldn’t have without this collaborative project.”
In applying the new CMINT algorithm, Sridharan and Roy have already found some surprising things. In one study, Sridharan was converting mouse embryonic fibroblasts into induced pluripotent stem (iPS) cells, with an intermediate partially-reprogrammed cell type called pre-iPSC. Using CMINT, Sridharan and Roy found that in the pre-iPS state, changes in the chromatin consistent with fully-reprogrammed iPS cells – two specific histone modifications – had already occurred, but without the corresponding gene expression associated with iPS cells.
“This means that these two histone modifications are not sufficient to turn on gene expression,” explains Sridharan, “but then why are these two even present? Do they set up the platform for other [modifications] to come in?” Sridharan hopes to learn more about what she calls poised states, where changes in chromatin have occurred but without large changes in gene expression. “Those are the kinds of follow-up experiments that are very interesting,” she says “which we could not have done without knowing this pattern.”
CMINT chart
Roy adds, “this is for a gene which is already known to be important, but those transitions can then be used to look for other new genes. It can be used for a hypothesis generation tool to do new experiments that could potentially help with understanding the biology of the system.” Furthermore, Sridharan may be able to use CMINT to identify specific genes to perturb in the reprogramming process, making development of iPS cells more efficient.
The method is not limited to “simple” systems with just three cell types like the mouse embryonic to pre-IPS to IPS system; the algorithm is flexible. To demonstrate its flexibility, Sridharan and Roy applied CMINT to another research group’s data including 15 different cell types in the hematopoietic system with histone modifications measured all over the genome. “It’s versatile in the kinds of things that you can use as input, and you can vary the number of cell types or the number of modifications; that’s another advantage of the tool,” says Sridharan.
Looking to the future, Sridharan and Roy want to improve the algorithm by adding information about another key player in gene expression: transcription factors, which are DNA-binding proteins that facilitate transcription and therefore gene expression. Data gathered in additional lab studies of transcription factors could be incorporated into CMINT to give scientists an even more complete picture of the gene regulatory networks associated with cell type transitions.
The CMINT software is available online, and may prove to be a powerful tool for discovering the key transition points in cell development. “It would be of interest to people who are looking at a dynamic process which they think is driven by chromatin state changes,” says Roy.
The paper, Chromatin module inference on cellular trajectories identifies key transition points and poised epigenetic states in diverse developmental processes, is in the July issue of Genome Research.
The work was supported in part by the National Institutes for Health (grants BD2K U54AI117924, R01GM117339 and R01GM113033) and the US Environmental Protection Agency (grant 83573701).
— Nolan Lendved
Related Posts


widiscoveryuw Instagram





