
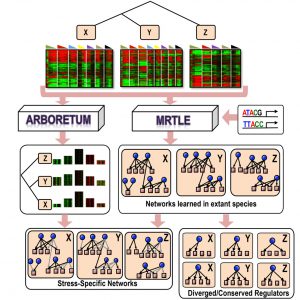
 Changes in transcriptional regulatory networks can significantly contribute to species evolution and adaptation. However, identification of genome-scale regulatory networks is an open challenge, especially in non-model organisms. Here, we introduce multi-species regulatory network learning (MRTLE), a computational approach that uses phylogenetic structure, sequence-specific motifs, and transcriptomic data, to infer the regulatory networks in different species. Using simulated data from known networks and transcriptomic data from six divergent yeasts, we demonstrate that MRTLE predicts networks with greater accuracy than existing methods because it incorporates phylogenetic information. We used MRTLE to infer the structure of the transcriptional networks that control the osmotic stress responses of divergent, non-model yeast species and then validated our predictions experimentally. Interrogating these networks reveals that gene duplication promotes network divergence across evolution. Taken together, our approach facilitates study of regulatory network evolutionary dynamics across multiple poorly studied species.
Changes in transcriptional regulatory networks can significantly contribute to species evolution and adaptation. However, identification of genome-scale regulatory networks is an open challenge, especially in non-model organisms. Here, we introduce multi-species regulatory network learning (MRTLE), a computational approach that uses phylogenetic structure, sequence-specific motifs, and transcriptomic data, to infer the regulatory networks in different species. Using simulated data from known networks and transcriptomic data from six divergent yeasts, we demonstrate that MRTLE predicts networks with greater accuracy than existing methods because it incorporates phylogenetic information. We used MRTLE to infer the structure of the transcriptional networks that control the osmotic stress responses of divergent, non-model yeast species and then validated our predictions experimentally. Interrogating these networks reveals that gene duplication promotes network divergence across evolution. Taken together, our approach facilitates study of regulatory network evolutionary dynamics across multiple poorly studied species.
Read the full paper in Cell Systems.
You must be logged in to post a comment.