Researchers Design New Strategy for Gene Therapy Development
The Saha Lab at the Wisconsin Institute for Discovery, in an article in Nature Communications, reports on an innovative combination of experiments and computation that can be used to develop new strategies for editing genes associated with genetic disorders. Their promising platform advances the CRISPR genome editing field and could lead to effective treatments for many diseases.
In the first few months of life, an infant with Pompe disease becomes weaker and weaker as glycogen, a complex sugar, builds up in their muscles, their cells unable to break it down. Multiple mutations in a gene called GAA prevent their cells from correctly producing the proteins needed to make lysosomes, the cells’ “food processors” that turn glycogen into glucose, the fuel that powers cells. Left untreated, most patients with Pompe die within a year.
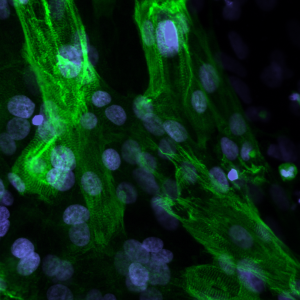
Bands of muscle in a cardiac cell derived from Pompe disease patient cells
The mutations that cause Pompe disease — and many other inherited diseases — are autosomal recessive, which means that they occur in both alleles (the genes inherited from both the mother and the father). Developing efficacious therapies for such diseases can be difficult for a number of reasons: first, diseases like Pompe have no animal models in which to test treatments, a typical step in therapy development. Further complicating the matter is the fact that many diseases are polygenic — resulting from mutations in two or more genes. The treatments themselves are often limited to adding functional genetic material to cells or injecting a functional protein product of GAA. These strategies have some promise, but do not repair the underlying mutations.
A new approach developed by Kris Saha, associate professor of biomedical engineering at the Wisconsin Institute for Discovery, uses precise gene editing tools to edit both faulty alleles simultaneously within individual cells to restore function. In an article published in Nature Communications on December 8, Saha’s lab describes how, absent an animal model exhibiting Pompe-similar mutations, the research team used induced pluripotent stem cells derived from Pompe patients to reproduce the exact GAA mutations that cause the disease and to approximate the resulting tissue pathology.
The lab successfully used a specially-designed, ultra-precise genome editing system to correct two distinct mutant alleles in an individual human cell. A previous study led by Jared Carlson-Stevermer, who was at the time a graduate student in Saha’s group, had established an up to 18-fold increase in precision of gene edits by combining a DNA repair template with the cutting machinery of CRISPR in one particle. But now, by using the method to fix two mutations at once in Pompe-derived cells, the researchers have been able to improve cell function dramatically, bringing lysosome protein production equivalent to the level of healthy cells without any major adverse effects that sometimes emerge from gene editing.

Krishanu Saha
But treating cells in the laboratory, while providing crucial insight, is the not the same as creating a therapy for patients. A critical step in developing treatments usually involves testing on animal models to evaluate efficacy and safety, a major obstacle for Pompe disease and other genetic conditions that lack viable animal models. “The exact mutations seen in the Pompe patients are not in an existing animal model, so we cannot do all of the preclinical studies that we would like to do to order to evaluate the safety and efficacy of different genome editing strategies,” says Saha.
To determine the best therapeutic strategy for polygenic diseases — evaluating different doses, delivery mechanisms and timing, risks, and other factors — the research team instead built a computational model that allows them to predict the outcomes of various conditions. “This allows us to survey a wider scope of many different gene therapies during the design of a strategy,” says coauthor Amritava Das, a postdoctoral associate at the Morgridge Institute for Research. “The computational approach is critical when you don’t have an animal model that resembles the human disease.”
After pumping close to a million simulation conditions through the computational model, Das, Carlson-Stevermer, and Saha have gained key insights about the delivery of gene editors into the livers of human infants with Pompe disease without having to subject a single patient to experimental treatments. And those insights establish that multiple-correction genome editing approach may be an effective treatment for Pompe and other polygenic recessive disorders.
The computational model, which can be easily adapted for other polygenic conditions, is a big step for the development of therapies for diseases like Pompe and lays the groundwork for a bridge from laboratory studies to the clinic. “We need a way to think about how we go from patient material to a therapy without having to build an animal model, a process that takes months to years and hundreds of thousands of dollars,” says Saha, who also notes that as more measurements are added to the model, it will gain more predictive power.
“It’s a very broad, adaptable platform,” Das says about the combined stem cell model and computational tool, “and a very different way of thinking about gene therapy.”
This work was supported by the National Science Foundation (CBET-1350178, CBET-1645123), the National Institutes for Health (1R35GM119644-01), the Environmental Protection Agency (EPA-G2013 –STAR-L1), the University of Wisconsin Carbone Cancer Center (P30 CA014520), the Wisconsin Alumni Research Foundation, and the Wisconsin Institute for Discovery.
Related Posts



widiscoveryuw Instagram





